

- Home
- Dipartimento
- Ricerca
- Didattica
- Post Lauream
- Trasferimento della conoscenza
- Come fare per





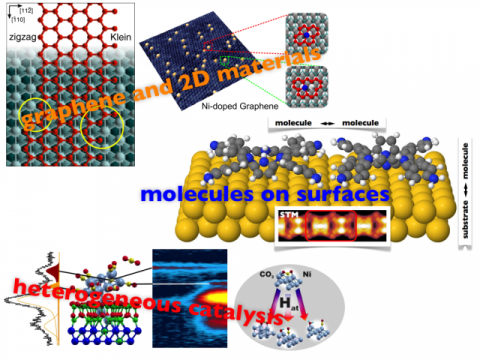
Struttura Elettronica dei Materiali: Teoria e Simulazione
Questo gruppo di ricerca è attivo nel campo della fisica teorica e computazionale della materia condensata a vari livelli.
I sistemi studiati sono principalmente: superfici, grafene e materiali bidimensionali, interfacce, etero e nanostrutture, adsorbati, catalisi eterogenea.
Il gruppo è coinvolto in esperimenti computazionali che fanno uso massiccio di simulazioni numeriche su scala atomica di materiali reali utilizzando principalmente un approccio basato sulla teoria del funzionale della densità (DFT) da principi primi. Al giorno d'oggi, queste simulazioni non solo rappresentano una chiave essenziale per interpretare i risultati sperimentali; grazie al loro potere predittivo sempre più affidabile, hanno una grande rilevanza dal punto di vista applicativo/tecnologico per la progettazione di nuovi materiali con opportune proprietà o di nuovi processi fisico/chimici.
Recentemente, uno sforzo particolare per superare i limiti dei metodi di struttura elettronica ab-initio è stato dedicato alla combinazione di questi con simulazioni atomistiche di dinamica molecolare su larga scala basate su tecniche sull'apprendimento automatico e sul calcolo ad alte prestazioni (HPC). Parte della ricerca prevede lo sviluppo di nuovi potenziali interatomici con tecniche di “machine learning” (apprendimento automatico) sui calcoli DFT, basati su reti neurali. L’automatizzazione del processo stesso di sviluppo di tali potenziali è oggetto di indagine, con l'obiettivo di impostare un flusso di lavoro standard per testare e validare i potenziali interatomici.
Una forte collaborazione anche con diversi gruppi sperimentali caratterizza da sempre questo gruppo di ricerca.
Info
Ultimo aggiornamento: 24-06-2024 - 18:25