

- Home
- Department
- Research
- Teaching
- Post Graduate Studies
- Knowledge Transfer
- How To





Electronic Structure of Materials: Theory and Simulation
Our field of interest is the computational physics of condensed matter at various levels.
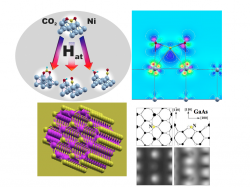 On the one hand, the group is involved in experiments making massive use of atomic-scale numerical simulations of real materials using a first principle approach. Nowadays, these simulations are not only an essential key for interpreting experimental results; thanks to their increasingly reliable predictive power, they are also of great help to technology for predicting the properties of materials and of physical/chemical processes, in the phases leading to the design of new materials or new processes. The systems studied are mainly: interfaces, surfaces, hetero- and nanostructures; semiconductors; semi-metals for spintronic applications; metallic surfaces, adsorbates, heterogeneous catalysis.
On the one hand, the group is involved in experiments making massive use of atomic-scale numerical simulations of real materials using a first principle approach. Nowadays, these simulations are not only an essential key for interpreting experimental results; thanks to their increasingly reliable predictive power, they are also of great help to technology for predicting the properties of materials and of physical/chemical processes, in the phases leading to the design of new materials or new processes. The systems studied are mainly: interfaces, surfaces, hetero- and nanostructures; semiconductors; semi-metals for spintronic applications; metallic surfaces, adsorbates, heterogeneous catalysis.
At the same time, important work is being done on developing new theories and models, as well as algorithms for an operative definition of some important properties and features of systems: piezoelectricity, ferroelectricity, flexoelectricity, theory of the insulating state, theory of polarization, theory of orbital magnetization.
For some subjects, such as, for instance, surface physics and heterogeneous catalysis, there is a strong collaboration with experimental groups, and in particular also of this Department and other local research Institutions.
Info
Last update: 04-08-2024 - 20:50